Whole genome sequencing reveals host factors underlying critical Covid-19 according to Nature (07 March 2022).
By Kousathanas, A., Pairo-Castineira, E., Rawlik, K. et al.
This study aims to compare the genomes from critically-ill cases with population controls in order to find underlying disease mechanisms. It is able with the GenOMICC study, the Genetics of Mortality in Critical Care.
There are sequenced 7 491 critically-ill cases compared with 48 400 controls. The comparison between critically-ill cases and population controls is highly efficient for detection of therapeutically-relevant mechanisms of disease. From this, it is discovered 23 independent variants that significantly predispose to critical Covid-19. There are identified independent associations with genes involved in the interferon signalling such as IL10RB (Interleukin 10 Receptor subunit Beta) and PLSCR1 (Phosophlipid Scamblase 1), both of them are protein coding genes. There are also genes involved in the leucocyte differentiation: BCL11A (BAF Chromatin Remodelling Complex Subunit BCL11A); and others involved in blood type antigen sectos status: FUT2 (Fucosyltransferase 2, mediates the inclusion of fucose in sugar moieties of glycoproteins and glycolipids).
Using a transcriptome-wide association study (TWAS), which detects associations between gene expression levels and phenotypic variation, the effect of gene expression on disease severity is transferred. In critical disease, expression of a membrane fippase ATPase Phospholipid Transporting 11A (ATP11A) is reduced, and mucin expression (MUC1) is increased.
Two distinct mechanisms can predispose to life-threatening disease: failure to control viral replication, or an enhanced tendency towards pulmonary inflammation and intravascular coagulation.
Since development of critical illness is in itself a key clinical endpoint for therapeutic trials, using critical illness as a phenotype in genetic studies enables detection of directly therapeutically-relevant genetic effects
There is previously discovered that critical Covid-19 is associated with genetic variation in the host immune response to viral infection and the inflammasome regulator DPP9 using microarrays. They are the ideal platform for assessing known markers in the human genome, enabling researchers to find single nucleotide polymorphisms (SNPs) or larger structural changes among millions of markers. DNA microarrays are used to measure the expression levels of large numbers of genes simultaneously or to genotype multiple regions of a genome.
GWAS analysis
They performed whole genome sequencing (WGS) to improve resolution and deepen fine-mapping of significant signals to enhance the biological insights into critical Covid-19. A genome-wide association study (GWAS) is used to identify genetic markers associated with phenotypic variation.
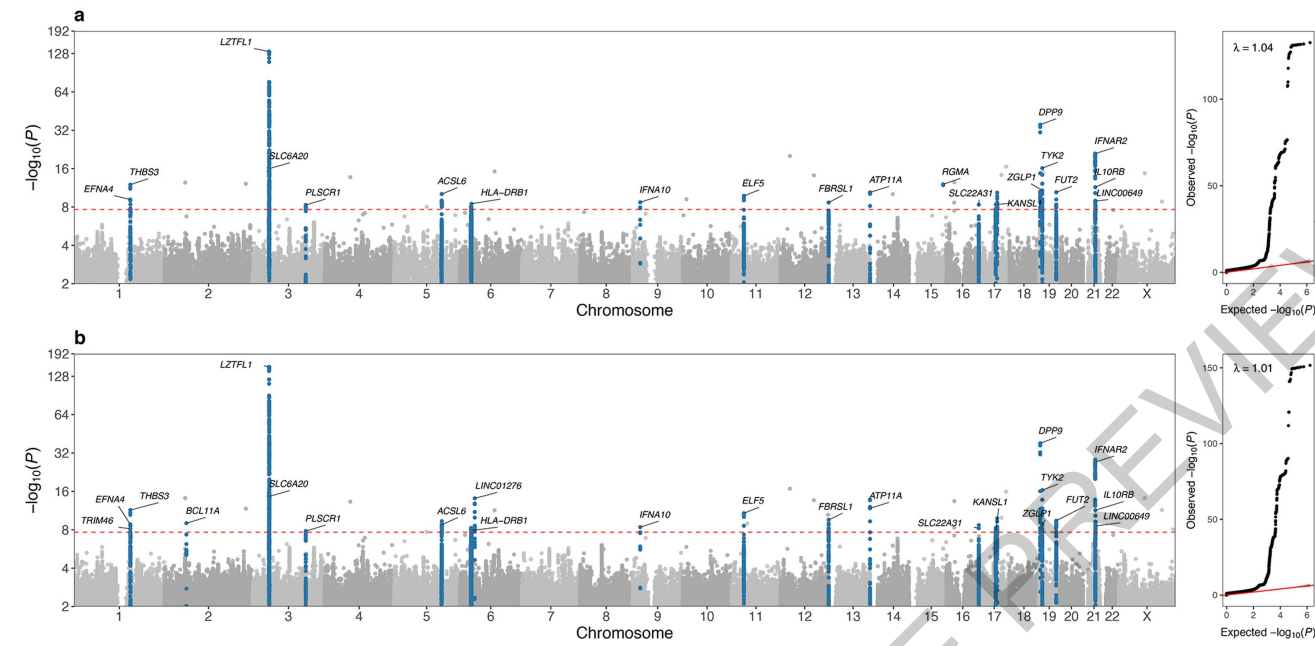
GWAS results for the EUR ancestry group. Results from a cohort of 7,491 critically-ill patients from 224 intensive care units, compared with 48,400 controls, describing discovery and validation of 23 gene loci for susceptibility to critical Covid-19.
To perform association analyses there is implemented SAIGE (Scalable and Accurate Implementation of Generalized mixed model). This method provides accurate P values even when case-control ratios are extremely unbalanced. SAIGE uses state-of-art optimization strategies to reduce computational costs; hence, it is applicable to GWAS for thousands of phenotypes by large biobanks.
There was required supporting evidence from variants in linkage disequilibrium for all genome-wide significant variants: observed z-scores for each variant were compared with imputed z-scores for the same variant, with discrepant values being excluded. A Z-score is a numerical measurement that describes a value's relationship to the mean of a group of values. Z-score is measured in terms of standard deviations from the mean.

Z-Score formula.
Since there is a theoretical risk of mismatch between cases and 100k participants in risk factors for exposure or susceptibility to critical Covid-19, there was performed a sensitivity analysis using only the mild cohort. In both of these analyses, allele frequencies and directions of effect were concordant for all lead signals
We inferred credible sets of variants using Bayesian fine-mapping with susieR (Sum of Single Effects Linear Regression). The methods are motivated by genetic fine-mapping applications, and are particularly well-suited to settings where variables are highly correlated and detectable effects are sparse.
They were able to fine-map multiple independent signals at previously identified loci. The signal in the 3p21.31 region (chromosome 3 p-arm band 21 and sub-band 31) was fine-mapped into two independent associations, with the credible set for the first refined to a single variant in the 5’ UTR region of SLC6A20 (chr3:45796521:G:T, rs2271616, OR:1.29, 95%CI:1.21,1.37) and the second credible set including multiple variants in downstream and intronic regions of LZTFL1.
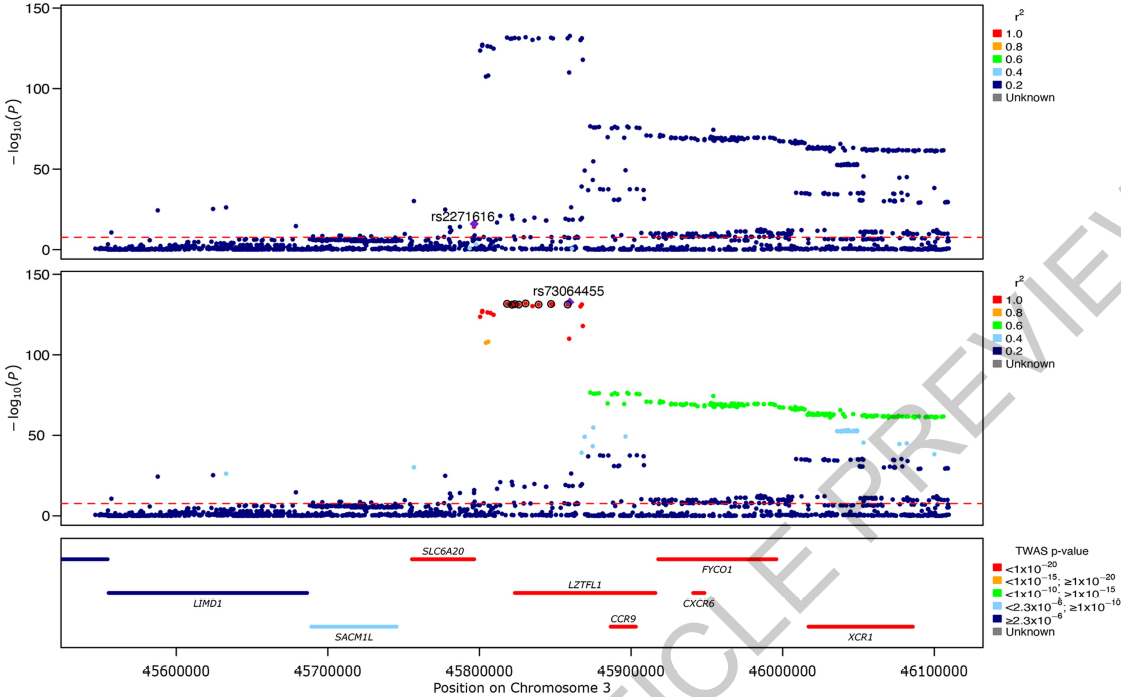
Regional detail showing fine-mapping to identify two adjacent independent signals on Chromosome 3.
Genetic susceptibility plays a stronger role in younger patients, age-stratified analysis in Europe revealed a signal in the 3p21.31 region with a significantly stronger effect in the younger age group. Sex-specific analysis did not reveal significant effects.
Replication
It is performed a meta-analysis of summary statistics generously shared by 23andMe, Inc. It is a publicly held personal genomics and biotechnology company based in Sunnyvale, California. Currently there are no comprehensive privacy regulations that would prevent governments from sharing DNA profiles with other groups, such as insurance companies.
There are replicated 23 of the 25 significant associations identified in the population specific and/or multi-ancestry GWAS. One of the non-replicated signals (rs4424872) corresponds to a rare variant that may not be well represented in the replication datasets.
HLA region
The HLA (Human Leukocyte Antigen) region lies on the short arm of chromosome six at position 6p21.3 (chromosome 6 p-arm band 21 and sub-band 3). The only allele that reached genome-wide significance was HLA-DRB1*04:01 (OutRadio C = 0.80, 95% 0I P .75 − 0.86, = 1.6 × 10−10 in Europe), which has a stronger P-value than the lead SNP in the region.
Transcriptome-wide association study (TWAS)
A transcriptome-wide association study (TWAS) detects associations between gene expression levels and phenotypic variation. In order to infer the effect of genetically-determined variation in gene expression on disease susceptibility, we performed a TWAS using gene expression data for two disease-relevant tissues, lung and whole blood. There are found significant associations between critical Covid-19 and predicted expression in lung and blood.
Mendelian randomisation
There is performed generalised summary-data-based Mendelian randomisation (GSMR). GSMR incorporates information from multiple independent SNPs and provides stronger evidence of a causal relationship than single SNP based approaches.
Of 16 proteome-wide significant associations in this study, 8 were replicated in an external dataset at a Bonferroni-corrected p-value threshold of P < 0.0031.
Discussion
There are reported 23 replicated genetic associations with critical Covid-19, discovered in only 7,491 cases. This demonstrates the efficiency of the design of the GenOMICC study, an open-source25 international research programme
Five critical Covid-19-associated variants have direct roles in interferon signalling and broadly concordant predicted biological effects. These include a probable destabilising amino acid substitution in a ligand, IFNA10 (Trp164Cys). The swapping of tryptophan for cysteine: the lack of cysteines resulted in structure destabilization and lack of tryptophan resulted in a less flexible peptide.
Finally, there was detected a lead risk variant in phospholipid scramblase 1 (nuclear localisation signal important for the antiviral effect of interferon). PLSCR1 controls replication of other RNA viruses.
Limitations
To minimise the risk of false positive associations due to technical artefacts, extensive quality measures were utilised.
Although we can have considerable confidence that the replicated associations with critical Covid-19 we report are robust, we cannot determine at which stage in the disease process, or in which tissue, the relevant biological mechanisms are active.
Conclusions
These genetic associations implicate new biological mechanisms underlying the development of life-threatening Covid-19, several of which may be amenable to therapeutic targeting. Even in the context of the ongoing global pandemic, translation to clinical practice is an urgent priority, biological and molecular studies, and, where appropriate, large-scale randomised trials, will be essential before translating our findings into clinical practice.