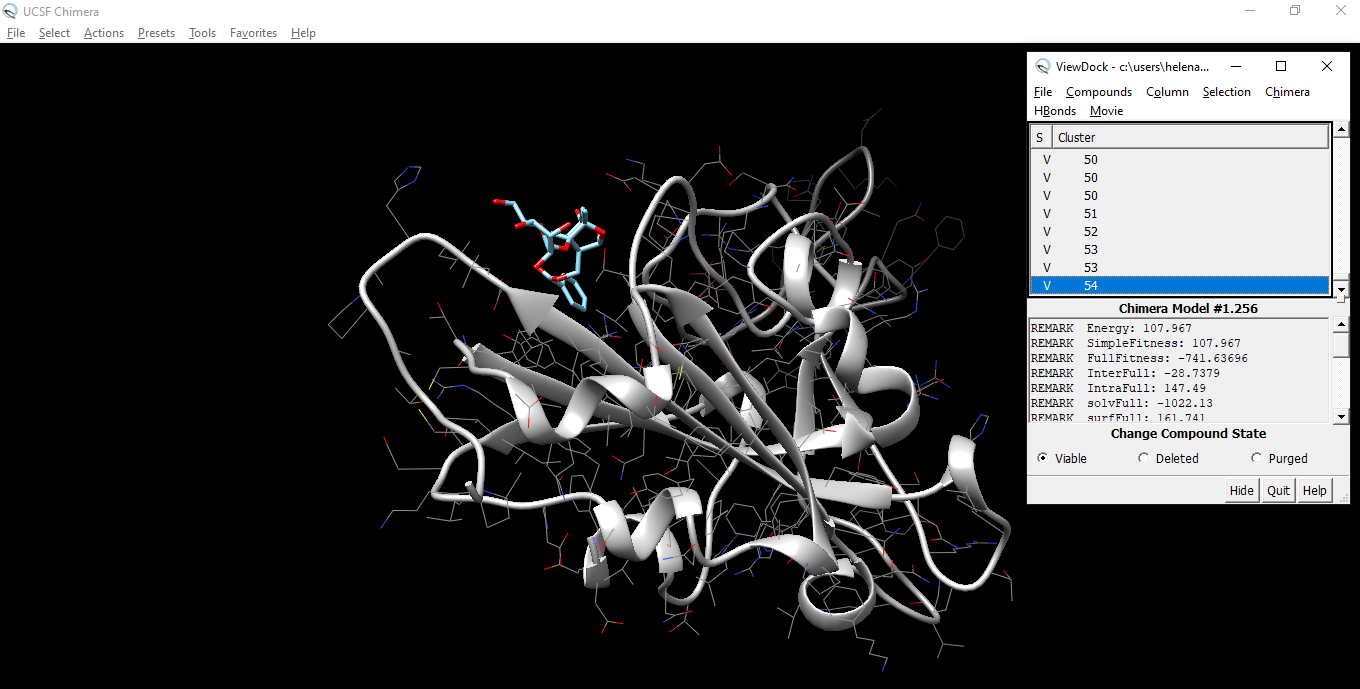
Isopaeniflorin
Once we have downloaded the Omicron spike (a glycoprotein that protrudes from the envelope of some viruses, such as a coronavirus, and facilitates entry of the virion into a host cell by binding to a receptor on the surface of a host cell followed by fusion of the viral and host cell membranes), we have to download an enzyme inhibitor (a molecule that binds to an enzyme and decreases its activity).
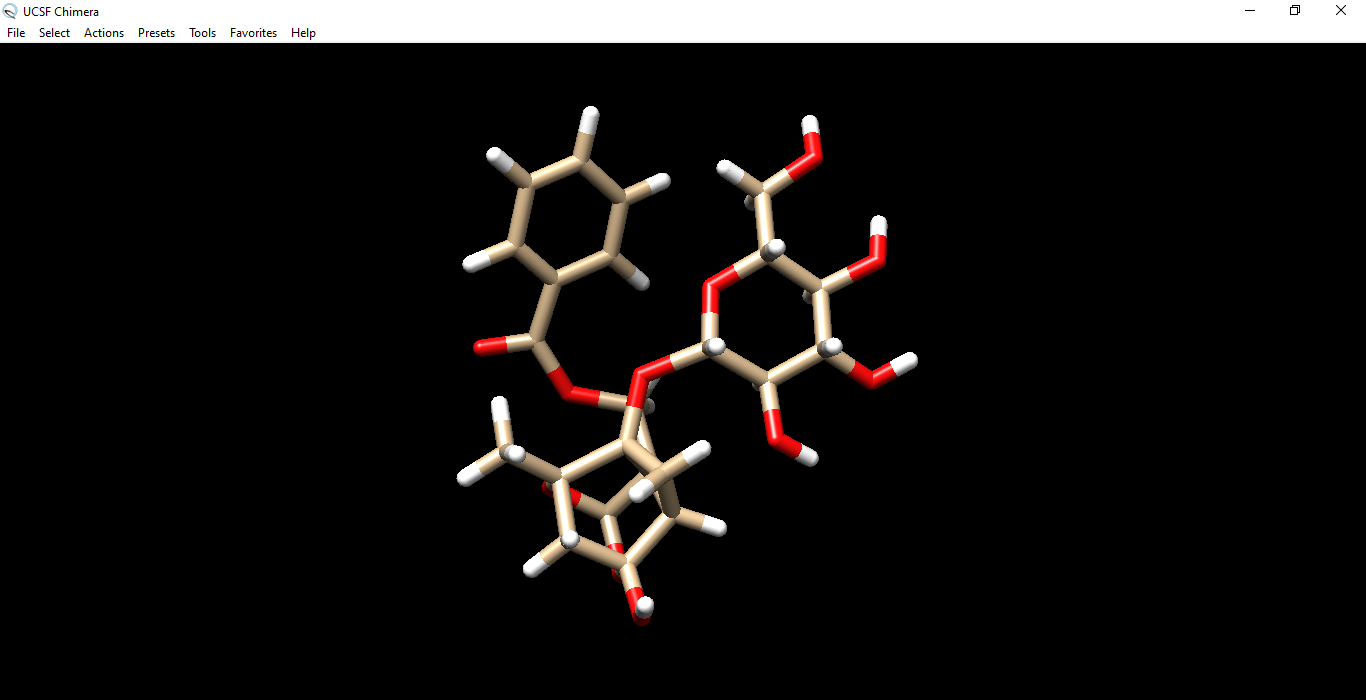
________________Omicron spike____________________________________________Isopaeniflorin inhibitor_____________
For doing the docking, in other words a molecular modeling technique that is used to predict how a protein (enzyme) interacts with small molecules (ligands), use Swissdock.
When we obtain the molecular docking, we open with Chimera the file titled as "open.chimerax". A batch can be made (a batch job is a program that is assigned to the computer to run without further user interaction), so drag the file to the application. Then, look for the molecule that has the most energy, because that means it is the one that inhibits the omicron spike the best. In the case of isopaeniflorin, this is the molecular docking at the omicron spike which has the higher energy.